

Designing complex molecules from the ground up represents one of the most formidable challenges in modern chemistry. It transcends mere knowledge of atomic connectivity; it demands an intricate understanding of reaction sequences, the strategic protection of sensitive molecular fragments, and the foresight to navigate away from potential pitfalls that could render months of meticulous laboratory work futile. Historically, this sophisticated expertise has resided predominantly within the minds of seasoned chemists, a testament to years of empirical learning and intuition. However, a groundbreaking development from the Swiss Federal Institute of Technology Lausanne (EPFL) is poised to democratize and accelerate this process, ushering in a new era of chemical discovery.
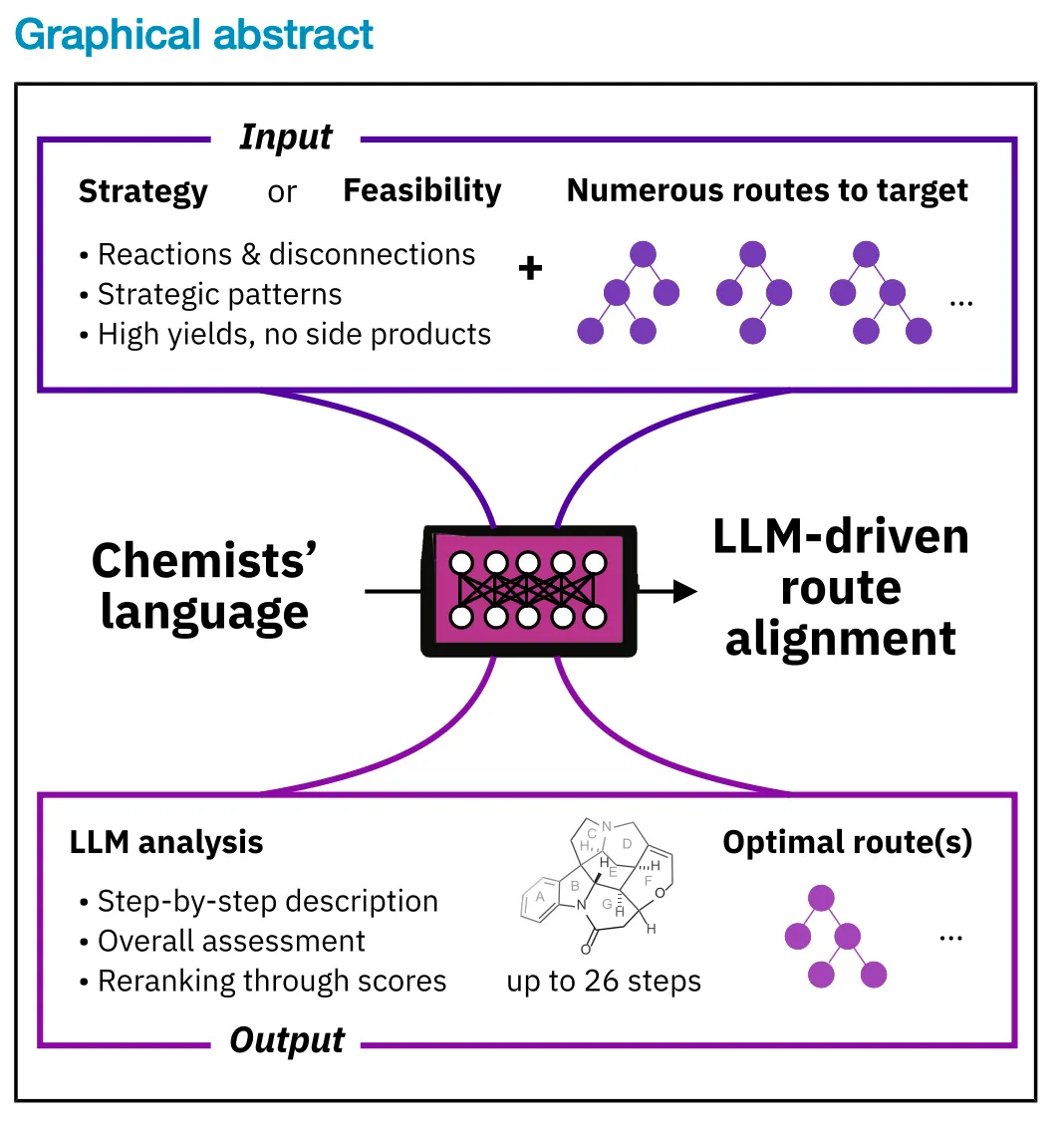
A team of researchers, spearheaded by Professor Philippe Schwaller, has unveiled Synthegy, an innovative framework that harnesses the power of large language models (LLMs) as sophisticated reasoning engines for chemical synthesis planning. Published this week in the prestigious journal Matter, the research introduces a subtle yet pivotal shift in how artificial intelligence is applied to chemistry. Instead of tasking AI with the generation of novel molecular structures, Synthegy leverages AI to meticulously evaluate and refine synthesis routes that are already generated by conventional software. This approach harnesses the predictive capabilities of LLMs to discern the most promising pathways among a multitude of possibilities, thereby streamlining the discovery and development of new chemical compounds.
The Synthegy Framework: A Novel Approach to Chemical Synthesis
The core innovation of Synthegy lies in its elegant workflow. A chemist initiates the process by inputting a desired molecular goal in plain, natural language. For instance, a request might be as specific as "form the pyrimidine ring in the early stages." This human-readable instruction is then fed into established retrosynthesis software. This type of software operates by deconstructing target molecules into progressively simpler precursor compounds, effectively working backward from the desired end product to identify potential starting materials and intermediate steps. The output of this traditional software is typically a vast array of potential synthesis routes, often numbering in the dozens or even hundreds, each representing a distinct sequence of chemical reactions.
Synthegy then ingeniously transforms each of these generated synthesis routes into a textual format. This textual representation is subsequently presented to a large language model. The LLM, acting as a sophisticated evaluator, meticulously scores each proposed route based on its congruence with the chemist’s initial English instruction. The most highly-rated routes, those that best align with the stated objective, are then surfaced to the top of the list. Crucially, Synthegy does not merely present a ranked list; it provides detailed written explanations accompanying each recommendation, elucidating precisely why a particular route is favored and how it fulfills the chemist’s requirements.

"When making tools for chemists, the user interface matters a lot, and previous tools relied on cumbersome filters and rules," stated Andres M. Bran, the lead author of the study, in a recent statement released by EPFL. This sentiment underscores a key driver behind the development of Synthegy: to create a more intuitive and accessible interface for complex chemical planning, moving beyond the often-arcane commands and parameters of legacy software.
Rigorous Validation and Performance Benchmarking
The efficacy of the Synthegy framework has been subjected to rigorous validation through a comprehensive double-blind study. This study involved 36 independent chemists, ranging from graduate students to senior researchers, who were tasked with evaluating 368 pairs of synthesis routes. The results were compelling: in 71.2% of cases, the chemists’ selections of the preferred route aligned precisely with the recommendations made by Synthegy. This figure is particularly noteworthy as it approximates the level of agreement typically observed among expert chemists themselves when assessing synthesis strategies, indicating that Synthegy is capturing the nuanced strategic thinking that underpins experienced chemical intuition.
Further analysis revealed an intriguing correlation between a chemist’s experience level and their agreement with Synthegy. Senior researchers, including professors and established research scientists, exhibited a higher degree of concordance with the AI’s suggestions compared to PhD students. This observation strongly suggests that Synthegy is adept at mirroring the sophisticated strategic intuitions and predictive capabilities that are honed through years of practical experience in the laboratory.
The researchers meticulously tested a variety of leading AI models within the Synthegy framework, including prominent LLMs such as GPT-4o, Claude, and DeepSeek-r1. The field of AI has been steadily advancing in drug discovery for years, with many existing approaches focusing on narrowly trained models designed for highly specific tasks. Synthegy, however, distinguishes itself through its modular design. This architectural flexibility allows it to seamlessly integrate with any existing retrosynthesis engine on the computational backend, while simultaneously leveraging the reasoning prowess of any capable LLM on the analytical side. In their benchmark tests, Google’s Gemini-2.5-pro emerged as the top-performing model, demonstrating exceptional accuracy and insight. Furthermore, DeepSeek-r1 was identified as a robust open-source alternative, offering the significant advantage of being capable of running locally, thereby enhancing accessibility and potentially reducing reliance on cloud-based services.
Beyond Synthesis Planning: Reaction Mechanism Elucidation
Synthegy’s capabilities extend beyond mere synthesis route planning; it also addresses the equally critical challenge of reaction mechanism elucidation. This aspect of chemistry delves into the fundamental question of why a chemical reaction occurs, seeking to understand the intricate movements of electrons and the precise sequence of elementary steps that transform reactants into products. Synthegy tackles this by systematically breaking down chemical reactions into their constituent elementary moves. The LLM then evaluates each proposed elementary step for its chemical plausibility, assessing whether it aligns with established principles of organic chemistry. In tests involving relatively straightforward reaction types, such as nucleophilic substitutions, the leading AI models integrated into Synthegy achieved near-perfect accuracy in predicting the reaction mechanisms.
Implications and Future Directions
The potential applications of Synthegy are vast and far-reaching, promising to accelerate innovation across numerous scientific and industrial sectors. Drug discovery, a field that has already witnessed significant AI-driven advancements, stands to benefit immensely. AI has demonstrated its ability to predict the outcomes of cancer treatments, and the principles behind Synthegy could be applied to identify novel therapeutic targets and optimize drug synthesis pathways. Beyond pharmaceuticals, the framework is equally applicable to the design of advanced materials with tailored properties, the optimization of industrial chemical processes for greater efficiency and sustainability, and the development of new catalysts.
A practical consideration highlighted by the research is the cost-effectiveness of the Synthegy approach. Evaluating approximately 60 candidate synthesis routes using the framework takes roughly 12 minutes and incurs API fees in the range of $2 to $3. This economic efficiency, combined with the significant time savings, makes Synthegy a compelling tool for both academic research and industrial development.
Despite its impressive capabilities, the researchers acknowledge certain limitations inherent in current LLM technology. The paper notes that LLMs can occasionally misinterpret the direction of a chemical reaction when presented in its textual representation, leading to erroneous feasibility assessments. Furthermore, smaller AI models have demonstrated performance no better than random guessing in this context, underscoring the importance of utilizing powerful, well-trained LLMs. The complexity of tracking coherent synthesis routes also increases significantly with length; routes exceeding 20 steps present greater challenges for LLMs to manage and analyze effectively.
The commitment to open science is evident, as the researchers have made the Synthegy code and benchmark datasets publicly available on GitHub at github.com/schwallergroup/steer. This transparency is expected to foster further research, development, and adoption of the framework within the global scientific community.
The development of Synthegy marks a significant leap forward in the application of artificial intelligence to the intricate world of chemistry. By empowering chemists with an intelligent assistant capable of navigating the complexities of synthesis planning and mechanism elucidation, this EPFL-led initiative is poised to accelerate the pace of scientific discovery, leading to the creation of new medicines, materials, and technologies that can address some of society’s most pressing challenges. The successful integration of LLMs into the chemical synthesis workflow represents not just a technological advancement, but a fundamental shift in how chemists can conceptualize, design, and ultimately realize the molecules of the future.
